Case study
Last updated on 2023-01-24 | Edit this page
Estimated time 122 minutes
Overview
Questions
- How do I analyse peptide count data in
R?
Objectives
- Demonstrate some ways of analysing NGS peptide count data
See the answers to each question for solutions.
Peptide count data
To round things out, we’ll explore some simulated peptide library data using what we’ve learned so far. Please follow along in an R markdown notebook on your own computer. The data is included in the data files you downloaded at the start of the course.
In this dataset, we’ve made an imaginary peptide insertion in the cap gene of AAV to create a plasmid library. We’ve then packaged this to create a vector library, and done a selection in some imaginary cells. We’ve therefore got data from plasmid, vector, DNA/entry and cDNA/expression libraries.
Imagine you’ve already done the counting using the counting pipeline, and now you want to explore the results.
You’ll start with four files, one for each library, each containing two columns: peptide and count. These files are called counts_plasmid.tsv, counts_vector.tsv, counts_entry.tsv, counts_expression.tsv.
The data here is simulated, so don’t interpret the results! If you want to see how I simulated the data, you can check it out on github.
Excercies
OUTPUT
── Attaching packages ─────────────────────────────────────── tidyverse 1.3.2 ──
✔ ggplot2 3.4.0 ✔ purrr 1.0.1
✔ tibble 3.1.8 ✔ dplyr 1.0.10
✔ tidyr 1.2.1 ✔ stringr 1.4.1
✔ readr 2.1.3 ✔ forcats 0.5.2
── Conflicts ────────────────────────────────────────── tidyverse_conflicts() ──
✖ dplyr::filter() masks stats::filter()
✖ dplyr::lag() masks stats::lag()R
# file paths
files <- here::here("episodes", "data", c("counts_plasmid.tsv",
"counts_entry.tsv",
"counts_expression.tsv",
"counts_vector.tsv"))
# load data
counts <- read_tsv(files, col_types = "ci", id="file")
# create extra column
counts <- counts %>%
mutate(lib = stringr::str_extract(file, "plasmid|vector|entry|expression")) %>%
select(-file)
glimpse(counts)
OUTPUT
Rows: 40,000
Columns: 3
$ peptide <chr> "KGEWPFI", "SILPAEY", "EGSLHTV", "MYNQSEE", "HYMWLTD", "WNCCNI…
$ count <int> 4, 3, 2, 1, 4, 3, 5, 7, 3, 2, 1, 2, 4, 6, 2, 5, 3, 2, 1, 6, 3,…
$ lib <chr> "plasmid", "plasmid", "plasmid", "plasmid", "plasmid", "plasmi…R
counts %>%
group_by(lib) %>%
summarise(n_pept = n_distinct(peptide),
mean_count = mean(count),
median_count = median(count),
sd_count = sd(count),
lib_size = sum(count))
OUTPUT
# A tibble: 4 × 6
lib n_pept mean_count median_count sd_count lib_size
<chr> <int> <dbl> <dbl> <dbl> <int>
1 entry 10000 3.18 2 6.18 31775
2 expression 10000 2.24 1 9.56 22367
3 plasmid 10000 3.98 3 3.00 39796
4 vector 10000 4.76 3 4.95 47553R
counts %>%
group_by(lib) %>%
slice_max(order_by=count, n=10)
OUTPUT
# A tibble: 43 × 3
# Groups: lib [4]
peptide count lib
<chr> <int> <chr>
1 DSGFDYR 173 entry
2 YYRVNEQ 139 entry
3 TKIVCQG 130 entry
4 PHGMDPM 112 entry
5 EKMPHRS 101 entry
6 PSFVTLG 87 entry
7 TQCNAIG 87 entry
8 GMEVEPY 85 entry
9 AALHTQL 83 entry
10 NCAPHKG 76 entry
# … with 33 more rowsR
counts <- counts %>%
group_by(lib) %>%
mutate(frac = count/sum(count))
glimpse(counts)
OUTPUT
Rows: 40,000
Columns: 4
Groups: lib [4]
$ peptide <chr> "KGEWPFI", "SILPAEY", "EGSLHTV", "MYNQSEE", "HYMWLTD", "WNCCNI…
$ count <int> 4, 3, 2, 1, 4, 3, 5, 7, 3, 2, 1, 2, 4, 6, 2, 5, 3, 2, 1, 6, 3,…
$ lib <chr> "plasmid", "plasmid", "plasmid", "plasmid", "plasmid", "plasmi…
$ frac <dbl> 1.005126e-04, 7.538446e-05, 5.025631e-05, 2.512815e-05, 1.0051…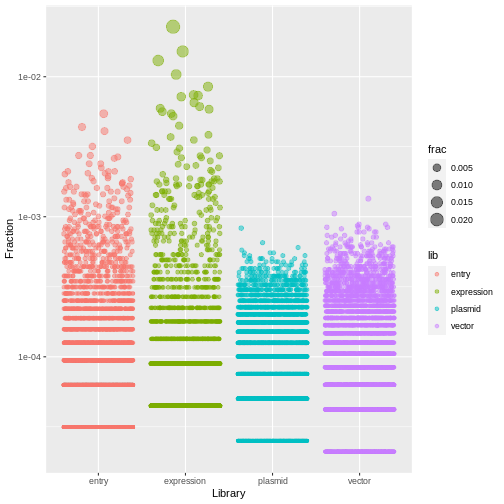
R
counts %>%
ggplot(aes(x=lib, y=frac, size=frac, color=lib)) +
geom_jitter(alpha=0.5, height=0) +
scale_y_log10() +
labs(x="Library", y="Fraction")
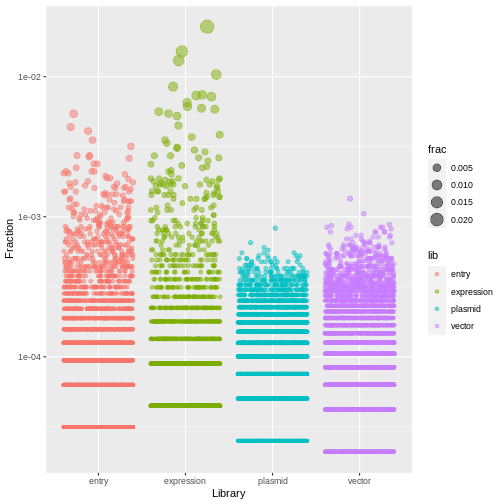
R
counts <- counts %>%
arrange(desc(frac)) %>%
group_by(lib) %>%
mutate(rank = row_number())
glimpse(counts)
OUTPUT
Rows: 40,000
Columns: 5
Groups: lib [4]
$ peptide <chr> "NKLAPNL", "DNPLDPY", "VEDTTFA", "HSCKITP", "RRRMETN", "DLGQGD…
$ count <int> 509, 339, 292, 232, 190, 166, 164, 161, 146, 137, 133, 131, 12…
$ lib <chr> "expression", "expression", "expression", "expression", "expre…
$ frac <dbl> 0.022756740, 0.015156257, 0.013054947, 0.010372424, 0.00849465…
$ rank <int> 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 1, 15, 16, 2, 3…Challenge
Make a table where there is one row for each peptide, and a column for the rank of the peptide in each library.
That is, there’s one column containing the counts for the plasmid library, one column for the counts in the vector library, and so on.
In this table, assign a unique ID (using row_number()) to each peptide, ranking by count in the expression library. That is, the peptide ranked 1 in the expression library should have ID 1, the peptide ranked 2 in the expression library should have ID 2, and so on.
Assign this new table to a variable ranked.
R
ranked <- counts %>%
select(lib, peptide, rank) %>%
pivot_wider(id_cols = "peptide", names_from="lib", values_from = "rank") %>%
arrange(expression) %>%
mutate(id = row_number())
ranked
OUTPUT
# A tibble: 10,000 × 6
peptide expression entry vector plasmid id
<chr> <int> <int> <int> <int> <int>
1 NKLAPNL 1 35 902 1807 1
2 DNPLDPY 2 80 171 396 2
3 VEDTTFA 3 92 189 301 3
4 HSCKITP 4 141 434 757 4
5 RRRMETN 5 186 541 909 5
6 DLGQGDE 6 226 766 2197 6
7 DLCLKKT 7 219 780 1930 7
8 PHGMDPM 8 4 131 266 8
9 CGHLPGF 9 288 1097 2934 9
10 HDIIKQH 10 275 1224 2754 10
# … with 9,990 more rows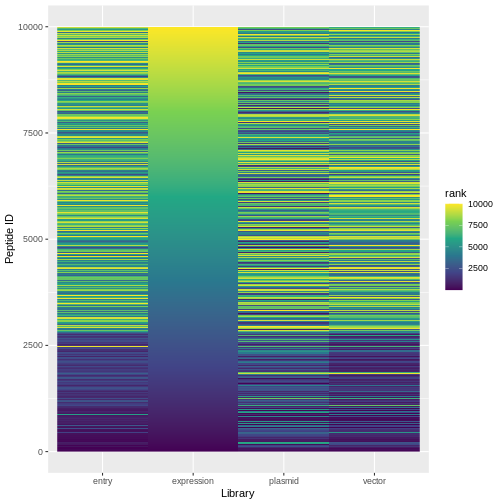
R
ranked %>%
pivot_longer(expression:plasmid, names_to="lib", values_to="rank") %>%
ggplot(aes(x=lib, y=id, fill=rank)) +
geom_raster() +
scale_fill_viridis_c()+
labs(x="Library", y="Peptide ID")
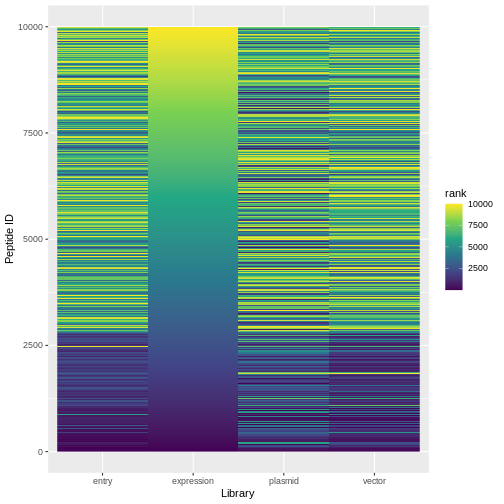
Challenge
Create a new table (called expr_enrich) in which you calculate the ‘enrichment score’ of each peptide in the expression library relative to the vector library (that is, \(\frac{\text{fraction in expression library}}{\text{fraction in vector library}}\)). Add a column called rank which ranks the peptides based on this enrichment score
R
expr_enrich <- counts %>%
filter(lib %in% c("expression", "vector")) %>%
select(peptide, lib, frac) %>%
pivot_wider(names_from = lib, values_from = frac) %>%
mutate(enrichment = expression / vector) %>%
arrange(desc(enrichment)) %>%
mutate(rank = row_number())
expr_enrich
OUTPUT
# A tibble: 10,000 × 5
peptide expression vector enrichment rank
<chr> <dbl> <dbl> <dbl> <int>
1 NKLAPNL 0.0228 0.000231 98.4 1
2 DNPLDPY 0.0152 0.000442 34.3 2
3 CGHLPGF 0.00653 0.000210 31.0 3
4 HSCKITP 0.0104 0.000336 30.8 4
5 VEDTTFA 0.0131 0.000442 29.6 5
6 DLGQGDE 0.00742 0.000252 29.4 6
7 HDIIKQH 0.00613 0.000210 29.1 7
8 DLCLKKT 0.00733 0.000252 29.1 8
9 RRRMETN 0.00849 0.000294 28.9 9
10 PGTAPIT 0.00300 0.000105 28.5 10
# … with 9,990 more rowsR
vec_enrich <- counts %>%
filter(lib %in% c("plasmid", "vector")) %>%
select(peptide, lib, frac) %>%
pivot_wider(names_from = lib, values_from = frac) %>%
mutate(enrichment = vector / plasmid) %>%
arrange(desc(enrichment)) %>%
mutate(rank = row_number())
poor_packagers <- vec_enrich %>%
filter(enrichment < 1)
poor_packagers
OUTPUT
# A tibble: 5,668 × 5
peptide vector plasmid enrichment rank
<chr> <dbl> <dbl> <dbl> <int>
1 MLCGFYC 0.0000421 0.0000503 0.837 4333
2 VGCWDVE 0.0000421 0.0000503 0.837 4334
3 KWVRLEQ 0.0000421 0.0000503 0.837 4335
4 YMRPDEV 0.0000421 0.0000503 0.837 4336
5 SFCASIY 0.0000421 0.0000503 0.837 4337
6 CPLGTGF 0.0000421 0.0000503 0.837 4338
7 PYNDHMH 0.0000421 0.0000503 0.837 4339
8 NFNNSQI 0.0000421 0.0000503 0.837 4340
9 ERVRIFK 0.0000421 0.0000503 0.837 4341
10 RFWAGYK 0.0000421 0.0000503 0.837 4342
# … with 5,658 more rowsR
good_peptides <- expr_enrich %>%
filter(!peptide %in% poor_packagers$peptide) %>%
slice_max(order_by=enrichment, n=500)
good_peptides
OUTPUT
# A tibble: 541 × 5
peptide expression vector enrichment rank
<chr> <dbl> <dbl> <dbl> <int>
1 NKLAPNL 0.0228 0.000231 98.4 1
2 DNPLDPY 0.0152 0.000442 34.3 2
3 CGHLPGF 0.00653 0.000210 31.0 3
4 HSCKITP 0.0104 0.000336 30.8 4
5 VEDTTFA 0.0131 0.000442 29.6 5
6 DLGQGDE 0.00742 0.000252 29.4 6
7 HDIIKQH 0.00613 0.000210 29.1 7
8 DLCLKKT 0.00733 0.000252 29.1 8
9 RRRMETN 0.00849 0.000294 28.9 9
10 PGTAPIT 0.00300 0.000105 28.5 10
# … with 531 more rowsR
write_tsv(good_peptides, file=here::here("good_peptides.tsv"))